
ODE model of actin polymerization and depolymerization with tracking of bound nucleotide
Description
This ordinary differential equation model describes actin polymerization and depolymerization with rates which depend on the hydrolysis state of bound nucleotide. The model accounts for the concentrations of monomeric actin bound to ATP, ADP-Pi, and ADP, and within filaments, the fraction of subunits at each location throughout the filament which are bound to ATP, ADP-Pi, or ADP. The identity of bound nucleotide is important for actin because it affects polymerization and depolymerization kinetics as well as the binding of several important binding proteins, including ADF/cofilin and Arp2/3 complex. The published model the current model was based on was the first to quantitatively identify the distribution of bound nucleotide along the length of actin filaments. All rate constants are adjustable, as are capping fractions for each end of the filament. The model output values include concentrations for all the nucleotide binding states for monomers, filament concentrations, and nucleotide binding state fractions for each individual subunit position throughout the simulated filaments. In addition, the fractions are assembled into profile variables in order to make it easier to view complete filament nucleotide profiles. These profile variables (Pt,Pdpi,Pd) are functions of two dimensions, s, which is subunit location within filaments measured from the barbed end, and t, time. So, a plot page curve of Pt(s,100) will show how the fraction of subunits bound to ATP varies along the entire filament after 100 seconds of simulation. A plot page curve of Pt(10,t), on the other hand, will show how the fraction of subunits bound to ATP at the 10th subunit from the barbed end varies over time. In the JSim realization of this model, we need a fixed number of equations and therefore need to fix the length of the filaments we are simulating. If you would like to investigate filaments of a different length than the default for this download, this is made easier by the use of the MPC utility (available at imagwiki.nibib.nih.gov/physiome/jsim/models/webmodel/NSR/MPC/). If you are interested in this option, make sure that you download the ActinCycle1.mpc file for this model as well as the ActinCycle1.proj file, since you will need the MPC source code to generate a new model file with the correct number of equations for your desired filament length. Detailed instructions can be found in the model source code and in the MPC source code. Note that doubling the length of the simulated filaments increases the simulation computation time by a factor of 5 to 6.
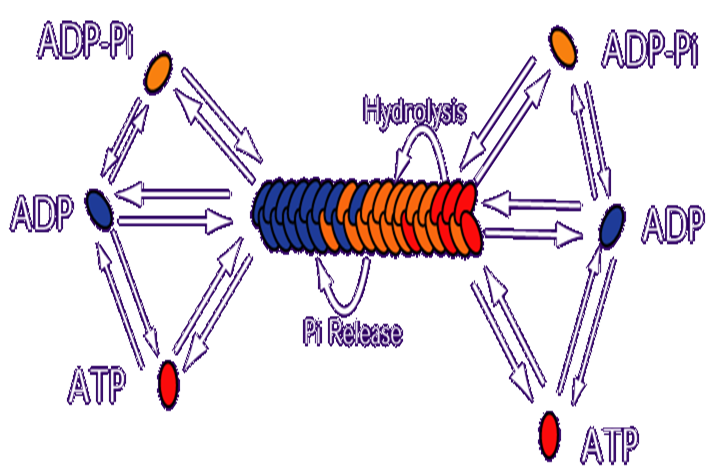
Each oval in the figure above represents an actin molecule. The color of the oval represents the state of the bound nucleotide. Red = ATP bound, Orange = ADP-Pi bound (where ATP hydrolysis has occurred, but the Phoshphate ion has not dissociated from the actin), Blue = ADP bound. On actin monomers, hydrolysis is negligible, so the primary methods of changing the state of the bound nucleotide are Pi release (which converts a monomer from ADP-Pi bound to ADP bound) and nucleotide exchange, where an actin monomer releases it's nucleotide altogether and binds a new one. In our model, we assume an excess concentration of free ATP in solution, so nucleotide exchange results in an ATP bound monomer. Monomeric actin molecules polymerize to form filaments. Filaments have two kinetically distinct ends and all lengthening or shortening of filaments occurs from subunits associating or dissociating from one of these ends. Both association and dissociation rate constants tend to be higher at one end of the filament, referred to as the "barbed" end. The other end is referred to as the "pointed" end. Once incorporated into filaments, ATP-bound actin subunits convert to ADP-Pi bound actin subunits through hydrolyzing their ATP, and ADP-Pi subunits convert to ADP-bound subunits by releasing their Pi. The ratio of on to off rates is higher at the barbed end than the pointed end, which results in this system settling into a steady state where there is net polymerization of ATP-actin at the barbed end and net depolymerization of ADP-actin at the pointed end, meaning that there is a flux of subunits moving from the barbed to the pointed end. This "treadmilling" is indirectly powered by the ATP being drawn from the surrounding solution.
Equations
ODEs governing changes in monomer concentrations:
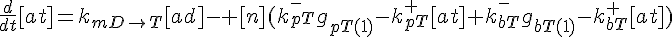
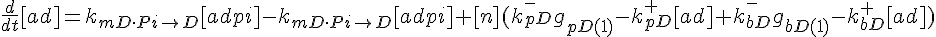
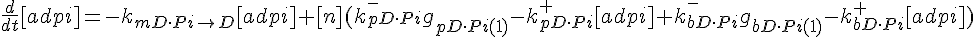
Flow into and out of filamentous pool
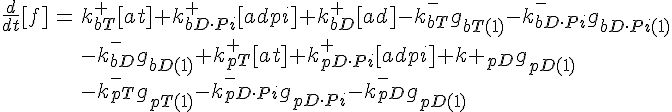
Conservation of Actin
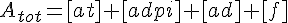
ODEs governing filament tip subunit fractions
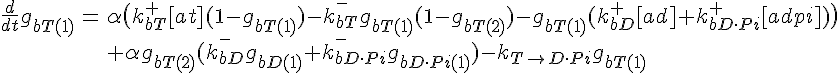
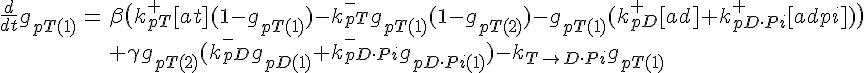
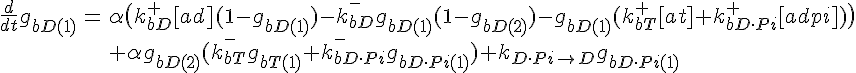
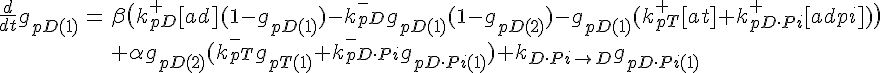
Filament tip subunit fractions must sum to one:
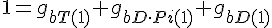
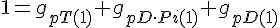
Equations governing internal subunit fractions
The following are the equations for the ith subunit from the tip, counting from the barbed end:
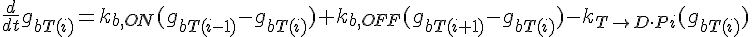
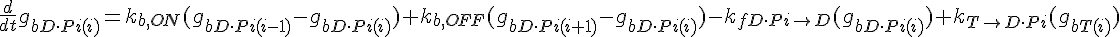
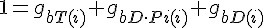
where 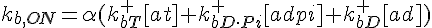 ,
, 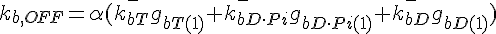
The following are the equations for the ith subunit from the tip, counting from the pointed end:
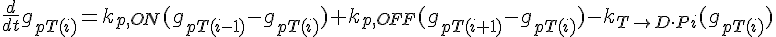
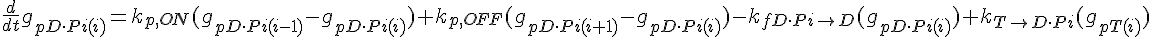
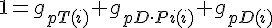
where 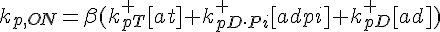 ,
, 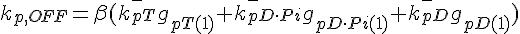
For the subunit at the exact center of the filament (designated here by location i*), we require that it lies on a cubic spline connecting subunits from the barbed and pointed end governed equations. This guarantees that the value of the profile and it's derivative vary smoothly across this center connection point.
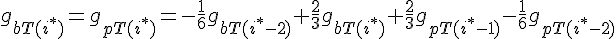
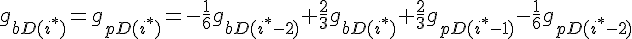
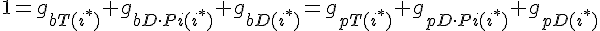
The equations for this model may be viewed by running the JSim model applet and clicking on the Source tab at the bottom left of JSim's Run Time graphical user interface. The equations are written in JSim's Mathematical Modeling Language (MML). See the Introduction to MML and the MML Reference Manual. Additional documentation for MML can be found by using the search option at the Physiome home page.
- Download JSim model MML code (text):
- Download translated SBML version of model (if available):
- No SBML translation currently available.
- Information on SBML conversion in JSim
We welcome comments and feedback for this model. Please use the button below to send comments:
Please cite https://www.imagwiki.nibib.nih.gov/physiome in any publication for which this software is used and send one reprint to the address given below:
The National Simulation Resource, Director J. B. Bassingthwaighte, Department of Bioengineering, University of Washington, Seattle WA 98195-5061.
Model development and archiving support at https://www.imagwiki.nibib.nih.gov/physiome provided by the following grants: NIH U01HL122199 Analyzing the Cardiac Power Grid, 09/15/2015 - 05/31/2020, NIH/NIBIB BE08407 Software Integration, JSim and SBW 6/1/09-5/31/13; NIH/NHLBI T15 HL88516-01 Modeling for Heart, Lung and Blood: From Cell to Organ, 4/1/07-3/31/11; NSF BES-0506477 Adaptive Multi-Scale Model Simulation, 8/15/05-7/31/08; NIH/NHLBI R01 HL073598 Core 3: 3D Imaging and Computer Modeling of the Respiratory Tract, 9/1/04-8/31/09; as well as prior support from NIH/NCRR P41 RR01243 Simulation Resource in Circulatory Mass Transport and Exchange, 12/1/1980-11/30/01 and NIH/NIBIB R01 EB001973 JSim: A Simulation Analysis Platform, 3/1/02-2/28/07.